29.03.2024
18.01.2024
La constitution d’un dossier d’AMM
En Europe, l’accès au marché des médicaments à usage humain est réglementé par la directive 2001/83/CE, transposée en droit français dans le Code de la Santé Publique (CSP). Ainsi, toutes les spécialités pharmaceutiques doivent disposer d’une Autorisation de Mise sur le Marché (AMM) avant de pouvoir être commercialisées. Qu’est-ce qu’une AMM ? Comment l’obtenir ? Manon, Chargée d’Affaires Réglementaires, nous informe sur le sujet !
4 minutes de lecture
La constitution d’un dossier d’AMM en Europe pour un médicament à usage humain
Qu’est-ce qu’une AMM ? [1]
Tout laboratoire pharmaceutique a pour obligation d’obtenir une Autorisation de Mise sur le Marché (AMM) afin de pouvoir commercialiser un médicament (art.6, directive 2001/83/CE). En France, l’AMM est délivrée par l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM).
L’AMM est un document de quelques pages, constitué d’une décision d’octroi d’AMM et d’annexes (Résumé des Caractéristiques du Produit (RCP), étiquetage, notice).
La délivrance d’une AMM est fondée sur l’examen de la balance bénéfice/risque du produit. Aussi, le laboratoire pharmaceutique doit fournir un ensemble de données scientifiques qui seront évaluées par une autorité compétente, nationale ou européenne. L’objectif est de démontrer que la qualité du médicament est maîtrisée, et qu’il est sûr et efficace pour le patient, dans des conditions d’utilisations précises.
Que contient un dossier d’AMM ? [2]
Le dossier d’AMM complet se présente sous un format standardisé : le CTD, pour Common Technical Document, ou e-CTD pour sa version électronique.
Ce format CTD a été développé par l’ICH (International Conference of Harmonisation), une organisation internationale rassemblant autorités de santés et professionnels de l’industrie pharmaceutique dans le but de faciliter l’harmonisation des données pharmaceutiques, notamment via la publication de guidelines et de standards.
Le format CTD est obligatoire en Europe et au Japon depuis 2003, et fortement recommandé aux Etats-Unis.
Le dossier d’AMM est organisé en 5 modules souvent représenté sous la forme du triangle CTD :
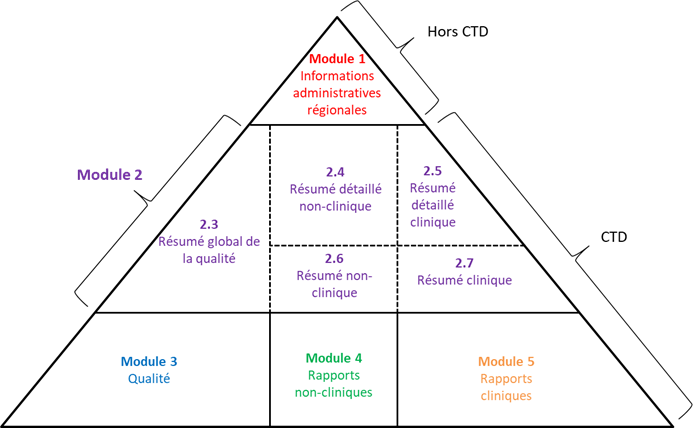
Le Module 1 est la partie administrative spécifique à chaque pays (hors CTD). Elle contient par exemple la lettre et le formulaire de demande, des informations sur le fabricant, le RCP, la notice, et l’étiquetage, la maquette des articles de conditionnement du médicament, les informations sur les experts, sur la pharmacovigilance, et l’évaluation du risque pour l’environnement.
Le Module 2 consiste en la synthèse des données chimiques, pharmaceutiques, non cliniques et cliniques, qui sont présentées et évaluées de manière critique.
Le Module 3 (« Quality », aussi connu sous le nom de partie CMC pour Chemistry, Manufacturing and Control), contient les informations chimiques, pharmaceutiques et biologiques sur le(s) principe(s) actif(s) et le produit fini, portant par exemple sur le développement, la fabrication, la caractérisation, le contrôle, le conditionnement, et la stabilité.
Le Module 4 (« Safety ») regroupe les rapports des différents essais non-cliniques in vitro et sur l’animal : rapports d’études pharmacologiques, pharmacodynamiques, pharmacocinétiques et toxicologiques.
Le Module 5 (« Efficacy ») regroupe les rapports des essais cliniques : rapports d’études biopharmaceutiques, pharmacocinétiques utilisant des biomatériaux humains, pharmacocinétiques chez l’homme, de pharmacodynamie chez l’homme, d’efficacité et de sécurité, sur l’expérience après mise sur le marché.
Remarque : dans certains cas particuliers (médicaments génériques, biosimilaires, hybrides, usage bien établi …), le dossier de soumission peut être simplifié, et les Modules 4 et 5 contenir des éléments bibliographiques. [3]
Un dossier d’AMM est une compilation de nombreux documents, nécessitant plusieurs expertises dans différents domaines. Sa constitution est longue, et sa rédaction peut commencer très en amont dans le cycle de développement du médicament, lors des essais pré-cliniques.
Comment obtient-on une AMM ? [1,4]
Il existe 4 procédures permettant d’obtenir une autorisation de mise sur le marché (AMM) : la procédure nationale et 3 procédures européennes, la procédure centralisée (CP), la procédure décentralisée (DCP) et la procédure par reconnaissance mutuelle (MRP). Chaque procédure a ses particularités et un calendrier qui lui est propre.
Le schéma ci-dessous présente les différentes étapes du traitement des demandes d’AMM en procédure nationale en France :
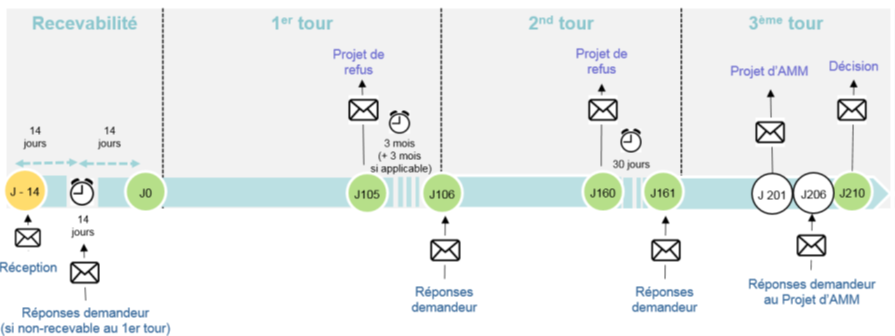
Dans un premier temps, le demandeur peut informer l’autorité de santé de sa démarche. Le demandeur va ensuite préparer le dossier de demande au format e-CTD, et le soumettre via la plate-forme de soumission dédiée, ici le CESP (Common European Submission Portal). La recevabilité du dossier va être étudiée. Lorsque le dossier est considéré comme complet et conforme, la procédure démarre : c’est le Jour 0 (J0). La phase d’évaluation dure 210 jours : elle porte sur les données scientifiques techniques et règlementaires fournies à l’appui de la demande ainsi que sur les propositions d’annexes de l’AMM (RCP, notice et étiquetage). Pendant l’évaluation, l’autorité peut demander des compléments d’information (J105 ; J160) : l’horloge s’arrête afin que le demandeur puisse fournir les réponses aux questions posées. Si le demandeur ne répond pas dans les délais impartis, ou si les éléments fournis demeurent insuffisants pour permettre l’octroi de l’AMM, la demande sera refusée. Si l’issue de l’évaluation est positive, un projet d’AMM sera envoyé par l’ANSM au demandeur (J201). Celui-ci disposera de 5 jours ouvrés pour vérifier les termes de la future AMM (J206). Finalement, l’autorité compétente prend sa décision (J210) et en informe le demandeur. La notification de la décision d’AMM et de ses annexes est réalisée par courrier et les annexes de l’AMM sont envoyées en parallèle par mail.
Globalement, l’évaluation d’un nouveau médicament dure environ 1 an.
Et après avoir obtenu l’AMM ? [1]
L’AMM est délivrée pour une durée initiale de 5 ans. Elle sera ensuite à renouveler une fois sans limitation de durée (hormis exception : pour des raisons de pharmacovigilance, il peut y avoir un 2ème renouvellement nécessaire après 5 ans).
Toutefois, si le laboratoire décide de ne pas commercialiser le médicament, alors son AMM deviendra caduque après 3 ans.
Après obtention, l’AMM peut faire l’objet de modifications, à l’initiative du laboratoire (par exemple liées à un changement de composition, à l’ajout d’une taille de lot, à une extension d’indication…) ou imposées par les autorités compétentes (par exemple en vue de l’introduction d’un nouvel effet indésirable). Le laboratoire doit obtenir l’autorisation de mettre en œuvre ces changements, via, par exemple, le dépôt de demandes de variations.
Sources :
[1] ANSM. Autorisation de mise sur le marché pour les médicaments [en ligne]. Disponible sur : https://ansm.sante.fr/page/autorisation-de-mise-sur-le-marche-pour-les-medicaments (consulté le 06/11/2023)
[2] ICH. CTD [en ligne]. Disponible sur : https://www.ich.org/page/ctd (consulté le 06/11/2023)
[3] EMA. Generic and hybrid applications [en ligne]. Disponible sur : https://www.ema.europa.eu/en/human-regulatory/marketing-authorisation/generic-medicines/generic-hybrid-applications (consulté le 06/11/2023)
[4] ANSM. AMM : cas général pour les nouvelles demandes d’AMM nationales [en ligne]. Disponible sur : https://ansm.sante.fr/page/amm-cas-general-pour-les-nouvelles-demandes-damm-nationales (consulté le 06/11/2023)